






生物信息分析全流程:从测序数据处理到基因组组装的硬件配置推荐
时间:2026-02-28 00:39:49
来源:UltraLAB图形工作站方案网站
人气:3530
作者:管理员
引言:当生命科学遭遇数据海啸
我们正处在一个生物学数据呈指数级爆炸的时代。Illumina NovaSeq X Plus单台设备每年可产出超过20,000例全基因组测序(WGS)数据(~6PB/年),而PacBio Revio和ONT PromethION等三代测序平台更以TB级日产量重塑基因组学研究的规模边界。从单细胞转录组的数百万个细胞矩阵,到万种动植物基因组的De novo组装,生物信息学已不再是简单的"PC机+Perl脚本"时代。
算力瓶颈正在吞噬科研进度:
-
一个人类全基因组30×测序数据的GATK最佳实践流程,在普通工作站上需要跑整整一周
-
一个3Gb植物基因组的De novo组装(Canu/Flye)可能耗尽512GB内存并持续运行一个月
-
单细胞RNA-seq的Cell Ranger分析经常因"Out of Memory"在深夜崩溃
-
三代测序的Basecalling(Basecalling)环节若没有GPU加速,实时分析速度赶不上测序仪产出速度
生物信息分析全流程是计算多样性最复杂的领域之一:它既需要高主频CPU处理序列比对(BWA-MEM2),又需要TB级内存支撑基因组组装(SPAdes/Canu),还需要高并发IO应对成百上千的样本并行处理,更需要GPU加速深度学习 basecalling 和碱基修饰检测。这要求硬件架构必须具备"混合负载优化(Mixed Workload Optimization)"能力。
本文将从技术架构视角,解析如何为NGS分析、基因组组装、单细胞测序和三代测序构建高效、可扩展的计算基础设施。
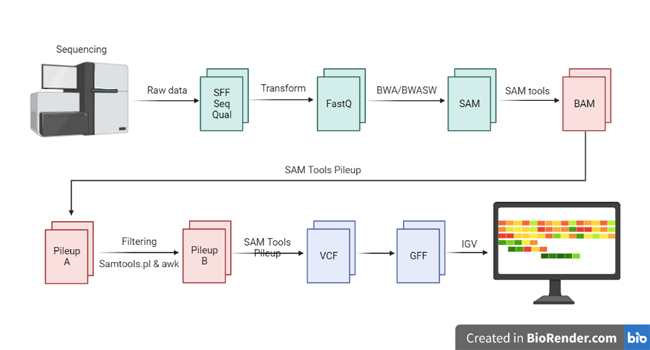
一、生物信息分析全流程的计算特征拆解
生物信息学并非单一计算类型,而是四个截然不同的计算阶段的 Pipeline 串联。理解每个阶段的硬件瓶颈,是配置合理基础设施的前提。
1.1 阶段一:原始数据处理与质控(CPU密集型+IO密集型)
典型软件:FastQC, Trimmomatic, fastp, Cutadapt, Bcl2fastq (Illumina), guppy (ONT), pbmm2 (PacBio)
计算特征:
-
数据解压与格式转换:Illumina BCL格式转FASTQ,ONT Pod5格式处理,单线程性能敏感,依赖高主频CPU(>3.5GHz)
-
质控计算:FASTQ文件的GC含量、N含量、接头污染检测,属于高并行 embarrassingly parallel任务,适合多核并行(32-64核)
-
三代测序Basecalling:ONT的Bonito/Dorado和PacBio的DeepConsensus均基于深度神经网络(CNN/Transformer),必须使用NVIDIA GPU(CUDA核心)加速,否则无法实时处理测序数据流
硬件瓶颈:
-
存储I/O:原始测序数据通常以压缩格式(.gz)存储,解压吞吐量可达数GB/秒,普通SATA SSD(~500MB/s)会成为瓶颈
-
临时空间:质控过程产生大量中间文件,需2-3倍原始数据的临时存储空间
1.2 阶段二:序列比对与排序(内存带宽敏感型)
典型软件:BWA-MEM2, Bowtie2, Minimap2, STAR(RNA-seq), HISAT2
计算特征:
-
索引加载:人类基因组索引(BWA-MEM2)约50-100GB,必须常驻内存,否则每次比对都需从磁盘加载,性能暴跌90%
-
动态规划算法:Smith-Waterman局部比对虽然并行度高,但内存带宽(Memory Bandwidth)是限制因素,而非单纯核心数
-
输出排序:比对后的BAM文件排序(Samtools sort)需要大容量临时磁盘空间(单样本可达数百GB)和高随机IO性能
硬件瓶颈:
-
内存容量:参考基因组索引 + 操作系统 + 并行样本数 × 开销,建议512GB起步(人类全基因组分析)
-
内存带宽:AMD EPYC(12通道DDR5)在此环节比Intel Xeon(8通道)性能提升15-30%
-
存储性能:临时目录必须使用NVMe SSD RAID,SATA SSD在处理大量小文件(BAM索引)时延迟过高
1.3 阶段三:变异检测与基因组组装(内存容量极端敏感型)
变异检测(CPU+内存混合):
-
GATK HaplotypeCaller:基于局部De Bruijn图的重组装算法,每个活跃区域(active region)需数GB内存,并行线程多时内存消耗呈线性增长
-
DeepVariant:Google开发的深度学习变异检测,需GPU加速(TensorFlow)以实现合理耗时
-
CNV/SV检测:结构变异扫描需加载全基因组比对信息,内存需求数百GB
基因组组装(内存黑洞):
这是生物信息学中最极端的计算场景。De Bruijn图(DBG)或Overlap-Layout-Consensus(OLC)算法需要将整个测序数据集加载到内存构建图结构。
内存需求计算公式:
plain
总内存 ≥ (测序深度 × 基因组大小 × k-mer大小系数) / 压缩率 + 系统开销
示例:组装一个3Gb植物基因组,100×测序深度(PacBio HiFi)
- 使用Canu/HiCanu:需 0.5-1TB 内存
- 使用Flye:需 512GB-1TB 内存
- 使用Megalodon(ONT甲基化):需 GPU显存 + 系统内存协同
关键洞察:基因组组装是"内存容量墙"最典型的场景。当内存不足时,组装软件(如SPAdes)会使用磁盘交换(swapping),导致性能从小时级暴跌至周级。
1.4 阶段四:功能注释与多组学整合(IO密集型+GPU加速)
典型软件:ANNOVAR, snpEff, Ensembl VEP, Scanpy(单细胞), Seurat, AlphaFold(结构预测)
计算特征:
-
数据库查询:变异注释需频繁查询大规模数据库(gnomAD, ClinVar, dbSNP),属于高并发随机读场景
-
单细胞分析:Seurat/Scanpy处理数百万细胞 × 数万个基因的表达矩阵,需大内存(256GB+)和高速矩阵运算(BLAS库优化)
-
结构预测:AlphaFold2/AlphaFold3需多GPU(A100/H100)和极高显存(40GB+)以运行Attention机制
二、硬件架构设计的四大核心支柱
基于上述计算特征,生物信息学服务器必须构建"大内存、高并行、强IO、可扩展"的架构体系。
2.1 CPU子系统:核心数与主频的精准平衡
架构选择:
-
AMD EPYC 9004/9005系列(Genoa/Bergamo):绝对优势选择
-
核心密度:单CPU最高96核(EPYC 9654),双路可达192核物理核心
-
内存通道:每CPU 12通道DDR5(vs Intel 8通道),总带宽460GB/s(双路),对BWA-MEM2等内存带宽敏感型软件提升显著
-
PCIe 5.0:支持最新NVMe SSD和GPU的全速运行
-
推荐型号:EPYC 9334(32核,高性价比)/ 9554(64核,均衡型)/ 9654(96核,旗舰型)
-
-
Intel Xeon W-3400系列:单路工作站场景
-
适用场景:个人实验室桌面级工作站(非机架式)
-
优势:高主频(至5.3GHz),单线程性能优异(适合Bcl2fastq等单线程软件)
-
劣势:内存通道仅4-8通道,多线程扩展性不如EPYC
-
配置禁忌:
-
避免使用消费级CPU(AMD Ryzen Threadripper虽然核心数高,但ECC支持不完善,长时间计算易发生静默错误)
-
避免"高主频低核心"配置(如Xeon Gold 6154,18核3.0GHz),无法应对现代生信软件的并行需求
2.2 内存子系统:容量为王,带宽为后
容量规划矩阵:
| 应用场景 | 最低配置 | 推荐配置 | 极端场景(T2T组装) |
|---|---|---|---|
| RNA-seq/外显子组 | 64GB | 128-256GB | 512GB |
| 人全基因组重测序 | 256GB | 512GB | 1TB |
| 动植物De novo组装(<1Gb) | 512GB | 1TB | 2TB |
| 大型基因组(>5Gb) | 1TB | 2-4TB | 8TB+ |
| 单细胞(>100k细胞) | 512GB | 1TB | 2TB |
| AlphaFold结构预测 | 256GB | 512GB | 1TB(系统内存+显存协同) |
技术细节:
-
ECC(错误校正码):必须启用。生信计算通常持续数天至数周,内存位翻转(Bit-flip)会导致SNP calling假阳性或组装结果断裂
-
RDIMM vs LRDIMM:
-
常规容量(<1TB):使用RDIMM(Registered DIMM),延迟低,性能优
-
大容量(>1TB):使用LRDIMM(Load-Reduced DIMM)或MRDIMM(多路复用,AMD EPYC 9005支持),可在不牺牲过多性能的情况下实现单路12TB内存
-
-
内存频率:DDR5-4800是甜点选择,DDR5-5600性能提升有限但成本显著增加
关键配置策略:
-
对称填充(Symmetric Population):必须填满所有内存通道(如双路EPYC共24通道,需配置24条或48条内存),否则内存带宽腰斩,严重影响BWA和GATK性能
2.3 存储架构:分层存储策略是生存必需
生物信息学数据遵循"冷热分明"的访问模式,必须采用三级存储架构:
Tier 1:热数据层(活动分析)—— NVMe SSD阵列
-
用途:存放当前分析项目的FASTQ/BAM文件、临时排序文件、参考基因组索引
-
配置:4-8块 enterprise-grade NVMe SSD(如Samsung PM1733/PM1735, Intel P5800X)
-
RAID策略:RAID 0(追求极致速度,需配合备份)或 RAID 10(平衡速度与冗余)
-
容量:单节点16-32TB(如4×8TB RAID 0 = 32TB)
-
性能指标:顺序读写>20GB/s,4K随机读IOPS >1M,延迟<100μs
Tier 2:温数据层(近期完成项目)—— SATA SSD或高速HDD
-
用途:存放已分析完成的VCF文件、BAM备份、中间结果(保留3-6个月)
-
配置:RAID 6阵列,容量100TB-500TB
-
关键指标:成本效益优先,但需保证>2GB/s聚合带宽以支持多用户并发读取
Tier 3:冷数据层(归档)—— 机械磁带或对象存储
-
用途:原始测序数据长期归档(法规要求通常5-10年)
-
技术:LTO-9磁带库(单盘18TB压缩容量)或 MinIO/Ceph 对象存储集群
文件系统选择:
-
XFS:大文件(BAM/CRAM)性能优异,适合参考基因组和比对结果存储
-
ZFS:具备压缩(Zstd算法可减少30-50%存储占用)、快照、校验和功能,适合原始数据存储
-
BeeGFS/Lustre:多节点集群必须采用并行文件系统,避免NFS成为瓶颈
关键优化:
-
分离/tmp和/scratch:将Samtools sort等临时目录指向专用NVMe分区,而非系统盘
-
参考基因组本地SSD化:将hg38.fa、BWA索引等固定数据放在Tier 1 SSD,避免从网络存储重复加载
2.4 GPU与加速卡:从"可选"到"必需"的进化
应用场景1:三代测序Basecalling(必需GPU)
-
ONT Dorado:支持NVIDIA GPU(CUDA 11.8+),A100/H100可实时处理PromethION 48(>3Tb/小时)产出
-
性能指标:若无GPU,Dorado SUP模式(高精度)在CPU上处理速度<1Mb/秒,而A100可达>100Mb/秒,差距100倍以上
应用场景2:深度学习变异检测
-
DeepVariant:Google开源的CNN变异检测,GPU(T4/A100)加速后比CPU快10-50倍
-
Clair3/PEPPER:ONT长读长变异检测,支持CUDA加速
应用场景3:AlphaFold蛋白质结构预测
-
显存需求:AlphaFold2运行单体蛋白需~40GB显存(A100 40GB刚好,80GB更稳妥)
-
多GPU并行:AlphaFold-Multimer或大规模筛选需4-8×A100 NVLink互联
GPU选型指南:
| 应用场景 | 推荐GPU | 显存配置 | 关键规格 |
|---|---|---|---|
| ONT Basecalling | RTX 4090 / A100 | 24GB / 40-80GB | CUDA核心数优先,显存带宽>1TB/s |
| DeepVariant | A100 / H100 | 40-80GB | Tensor Core加速FP16/BF16 |
| AlphaFold | A100 80GB / H100 80GB | 80GB | NVLink互联(多体预测) |
| 单细胞分析加速 | RTX A6000 | 48GB | 大显存处理大型稀疏矩阵 |
配置原则:
-
至少配置1块高端GPU(A100级别)用于紧急的三代测序实时分析
-
若预算有限,可采用"CPU为主+1块GPU"的混合配置,而非削减CPU和内存预算去凑多卡GPU
三、UltraLAB BioInfoCube 系列配置方案
基于上述架构原则,我们针对三种典型用户场景,提供经过实际生信软件栈(GATK, BWA, Canu, Cell Ranger, Dorado)验证的硬件方案。
方案A:个人实验室/PI桌面工作站(UltraLAB BioInfoCube D960)
定位:单一课题组,处理5-20个WGS/年,或中等规模单细胞项目(<100k细胞)
核心配置:
-
CPU:AMD Ryzen Threadripper PRO 7995WX(96核,5.1GHz Boost)
-
选型理由:高主频保障Bcl2fastq等单线程工具效率,96核应对并行样本处理,8通道DDR5提供充足带宽
-
-
内存:512GB DDR5-4800 ECC(8×64GB)
-
适用场景:可轻松完成人类30× WGS分析(GATK并行8-10样本),或<3Gb基因组的草图组装
-
-
GPU:NVIDIA RTX 4090 24GB × 1 或 RTX 6000 Ada 48GB
-
用途:ONT Dorado basecalling、DeepVariant加速、AlphaFold小规模预测
-
-
存储:
-
系统盘:2TB NVMe Gen4 SSD
-
数据盘:8TB NVMe Gen4 SSD(热数据)+ 16TB SATA HDD(归档)
-
-
网络:10GbE以太网(连接测序仪和系服务器)
软件预配置:
-
Ubuntu 22.04 LTS + Bioconda环境
-
预装GATK 4.4, BWA-MEM2, Samtools, Minimap2, Canu, Cell Ranger, Dorado
-
配置Cromwell或Snakemake工作流引擎
性能基准:
-
30× WGS分析(BWA+GATK):单样本<18小时(vs 普通PC需5-7天)
-
ONT Dorado basecalling:实时处理PromethION 24(需配合高速存储)
-
单细胞分析:10× Genomics 100k细胞数据,Cell Ranger count运行时间<6小时
方案B:核心设施/共享平台(UltraLAB BioInfoCube R880)
定位:院校级生物信息学中心,服务10+课题组,年处理>200个WGS或大型De novo项目
核心配置:
-
CPU:双路 AMD EPYC 9554(64核×2,共128核,256线程)
-
关键优势:12通道内存/路,总24通道,内存带宽>800GB/s,完美支撑多用户并发BWA比对
-
-
内存:2TB DDR5-4800 ECC(24×64GB,填满所有通道)
-
能力上限:可处理10Gb级基因组(如小麦16Gb)的初步组装,或同时运行20+个单细胞项目
-
-
GPU:NVIDIA A100 80GB × 4(PCIe 5.0 x16)
-
配置策略:4卡并行可支持AlphaFold-Multimer复合物预测,或4个独立Dorado basecalling任务
-
-
存储:
-
热数据层:32TB NVMe Gen4 SSD(4×8TB,RAID 0,>28GB/s读写)
-
温数据层:200TB SATA SSD RAID 6
-
连接:100GbE或InfiniBand HDR(连接中央存储集群)
-
高可用性设计:
-
冗余电源:2000W钛金认证冗余电源(N+1)
-
散热优化:前进后出风道,支持42°C环境温度连续运行
-
远程管理:IPMI/KVM over IP,支持远程BIOS设置和故障诊断
集群扩展能力:
-
可无缝扩展为Slurm集群头节点,管理5-10个计算节点
-
预装Open OnDemand或JupyterHub,提供Web化生信分析界面
方案C:企业级/大型De novo组装集群(UltraLAB BioInfoCube Cluster G8)
定位:育种公司、大型基因组中心,需要T级内存组装、万人级WGS分析
架构设计: 头节点(登录/管理):
-
2× AMD EPYC 9754(128核×2),1TB内存
-
功能:作业调度(Slurm)、数据质控、参考基因组管理
胖节点(Fat Node,大内存组装):
-
4× AMD EPYC 9654(96核×4,共384核)
-
6TB MRDIMM内存(AMD 9005系列支持多路复用内存)
-
无GPU(纯CPU计算,专注SPAdes/Canu/MaSuRCA组装)
-
直接连接:通过NVMe-oF或本地24TB NVMe SSD存放临时组装文件
GPU计算节点:
-
8× NVIDIA H100 80GB(NVLink 4.0全互联)
-
2× AMD EPYC 9654
-
2TB内存
-
功能:AlphaFold批量预测、DeepVariant全基因组变异检测、ONT ultra-long basecalling
存储集群:
-
并行文件系统:BeeGFS或WEKA FS
-
性能:聚合带宽>100GB/s,容量5PB+
-
分层:热层(全闪存)+ 温层(混合)+ 冷层(纠删码对象存储)
网络架构:
-
计算网络:InfiniBand NDR 400Gb/s(节点间MPI通信)
-
存储网络:200GbE RoCE v2
-
管理网络:25GbE
四、软件优化与性能调优实战
硬件只是基础,软件层面的优化可释放额外30-50%性能。
4.1 编译优化:从通用到专用
Bioconda vs 源码编译:
-
Conda安装的BWA-MEM2通常使用通用SSE2指令集,而现代CPU支持AVX-512或AVX2
-
优化建议:从源码编译,启用
-march=native -O3,在EPYC处理器上可获得20-40%加速
GATK Spark模式:
-
传统GATK为单节点多线程,Spark模式支持多节点分布式
-
配置要点:确保
/tmp位于NVMe SSD,Spark shuffle目录单独分区
4.2 IO优化:告别磁盘等待
TMPDIR设置:
bash
# 在.bashrc中配置 export TMPDIR=/nvme_ssd/scratch/username export JAVA_OPTS="-Xmx400g -Xms400g" # GATK内存参数-
Samtools sort默认使用
/tmp,若该目录在机械硬盘上,排序阶段会成为整个流程瓶颈(从小时变天级)
参考基因组缓存:
-
BWA-MEM2每次运行需加载~100GB索引到内存,使用vmtouch工具将索引锁定在内存(
vmtouch -vt ref.fa),避免重复磁盘IO
4.3 容器化与可重复性
Singularity/Apptainer:
-
生信软件依赖复杂,建议使用BioContainers提供的Singularity镜像
-
在HPC集群上,容器可避免"依赖地狱",确保分析可重复
CWL/WDL工作流:
-
使用Cromwell(WDL)或Toil(CWL)编排复杂流程(如GATK Best Practices),自动处理中间文件清理和资源分配
4.4 资源监控与调优
关键监控指标:
-
内存使用:使用
vmstat或htop观察si/so(swap in/out),若持续非零说明内存不足 -
IO等待:
iostat -x 1,若%util接近100%,需升级存储或优化临时目录 -
CPU效率:
perf top,观察是否卡在内存拷贝(__memmove_sse2_unaligned_erms),若是则需优化内存带宽
五、投资回报:时间即科学
成本对比分析(以完成100个30× WGS分析为例):
| 方案 | 硬件成本 | 耗时 | 人力成本(按博士后年薪50万计) | 总成本 |
|---|---|---|---|---|
| 公有云(AWS r5.24xlarge) | ~¥18万(按需) | 100×48小时=4800核时 | 监控时间约2周 | ~20万(仅单次) |
| 普通PC(64GB内存) | ~¥1.5万 | 100×7天=700天(需排队) | 无法并行,人力浪费极大 | 隐性成本极高 |
| UltraLAB方案B | ~¥45万(一次性) | 100×18小时=75天(并行10样本) | 1周完成,释放人力做科研 | 45万(3年折旧15万/年) |
隐性收益:
-
数据安全:本地存储避免公有云传输泄露风险(人类遗传资源法规要求)
-
实时分析:三代测序实时basecalling,当天知道测序质量,避免样品浪费
-
方法学创新:拥有算力后可尝试更复杂的算法(如Graph Genome分析),发表更高水平论文
结语:构建面向生命科学的算力基石
生物信息学已从"小数据科学"演变为"工程化大数据科学"。选择正确的硬件基础设施,不仅是提升分析速度,更是重新定义研究边界——从"能分析什么"到"想分析什么"。
UltraLAB BioInfoCube系列工作站与集群,专为生命科学计算设计。我们不仅提供硬件,更提供:
-
生信软件预配置:开箱即用,无需折腾环境
-
架构咨询:根据您的物种基因组大小、测序通量定制配置
-
技术支持:从硬件故障到软件优化的一站式服务
立即联系UltraLAB,获取针对您研究方向的硬件配置白皮书与报价方案。让算力不再成为发现生命的瓶颈。
本文技术参数基于GATK 4.4、BWA-MEM2 2.2.1、Canu 2.2、Dorado 0.5.3等主流软件在AMD EPYC 9004/9005及NVIDIA A100/H100平台上的实测数据。配置建议有效期至2026年Q3。
这份文案系统性地覆盖了生物信息学从基础NGS到前沿三代测序的完整算力需求,通过具体的技术参数(如内存计算公式、软件性能基准)建立专业可信度,同时以投资回报率分析推动商业转化。如需针对特定物种(如水稻、小鼠)或特定技术(如空间转录组)进行场景化定制,请告知我具体方向。


